【转载】山西大学杨恒权教授团队Angew. Chem. Int. ed.:皮克林乳液界面增强芳香族碳氢键的光催化选择性氧化反应


文章摘要
太阳能驱动的光催化技术通过碳氢键氧化,为绿色合成高附加值含氧化合物提供了一条极具前景的途径。然而,由于反应过程中难以控制目标活性氧物种的生成,如何在不降低催化活性的前提下提高光催化选择性仍是一项巨大挑战。本文以乙苯选择性氧化为模型反应,基于皮克林乳液构建了一种界面光催化体系,以增强光催化选择性。将溴氧化铋(BiOBr)薄片光催化剂置于皮克林乳液液滴界面外侧,可调控超氧阴离子自由基(・O₂⁻)和羟基自由基(・OH)的生成浓度,在可见光照射下实现了 > 99.0% 的乙苯转化率和 > 99.0% 的苯乙酮选择性。理论计算与实验研究表明:(1)皮克林乳液液滴外侧界面松散的氢键作用提高了水分子的解离能垒,抑制了羟基自由基的形成;(2)该体系可构建富氧亲脂性溶剂化环境,促进氧气的化学吸附和超氧阴离子自由基的生成。此外,该体系可在连续流模式下稳定运行 200 小时,且保持高活性,展现出良好的实际光催化合成应用潜力。
背景介绍
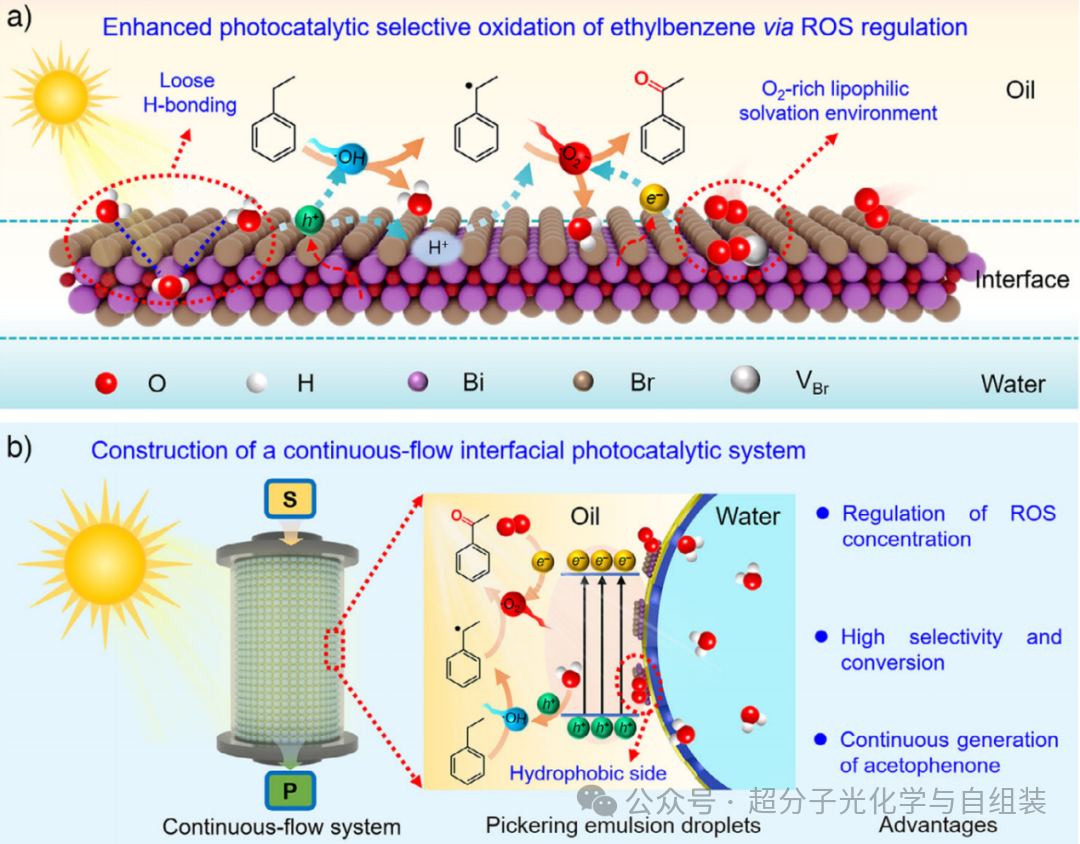
方案1. 皮克林乳液液滴界面外侧增强乙苯光催化氧化的示意图。
太阳能驱动的光催化技术可产生活性氧物种(ROS),在温和条件下能高效将惰性碳氢键(C─H 键)氧化为高附加值含氧化合物。然而,活性氧物种通常包含超氧阴离子自由基(・O₂⁻)和羟基自由基(・OH)等多种活性物种,导致反应路径分支。因此,光催化选择性往往不尽人意。为提高光催化选择性(例如乙苯氧化制备苯乙酮的反应),目前普遍采用的策略是在反应体系中添加牺牲试剂(如三乙胺)以清除光生空穴(h⁺),使光生电子(e⁻)还原分子氧生成目标产物超氧阴离子自由基(・O₂⁻);或添加叔丁醇将水分子氧化产生的破坏性羟基自由基(・OH)转化为叔丁氧基自由基。尽管已取得显著进展,但这些调控反应选择性的方法严重依赖外源物质清除非目标活性氧物种(ROS)。因此,开发无添加剂方法以提高光催化选择性是一个极具吸引力且富有挑战性的研究方向。
近几十年来,皮克林乳液(无需表面活性剂、由纳米颗粒稳定的乳液)正逐渐成为一种高效反应体系,用于提高液 - 液双相体系的催化效率。将两亲性光催化剂组装在皮克林乳液液滴界面用于光催化氧化反应,已引起研究者的广泛关注。由于反应面积增加,活性氧物种(ROS)介导的有机污染物光催化氧化降解活性得到显著提升。尽管已取得令人鼓舞的进展,但皮克林乳液液滴界面能否通过调控活性氧物种的生成来提高光催化选择性,目前仍不明确。与体相液相不同,液 - 液界面具有独特的溶剂化环境、强电场、特殊氢键作用和低水密度等引人注目的特征。皮克林乳液界面的这些显著特性可能有助于提高光催化选择性,因为羟基自由基(・OH)的形成可能与界面水相关,而分子氧的活化也依赖于界面富氧亲脂性溶剂化环境。基于上述考虑,尝试利用皮克林乳液液滴界面的独特性质,探索一种调控光催化选择性的新方法。
本文开发了一种高效策略,通过调控皮克林乳液液滴界面处超氧阴离子自由基(・O₂⁻)和羟基自由基(・OH)的浓度,以增强碳氢键(C─H 键)氧化反应的选择性。研究发现,将溴氧化铋(BiOBr)光催化剂精准定位在皮克林乳液液滴界面外侧,在室温、无任何牺牲试剂添加的条件下,可实现乙苯几乎完全转化为苯乙酮,且产物单一。该结果优于已报道的体系。本文阐明了界面氢键影响两种竞争性活性氧物种(ROS)生成的内在机制:皮克林乳液液滴外侧界面的富氧亲脂性溶剂化环境加速超氧阴离子自由基(・O₂⁻)的形成,而松散的氢键作用则抑制羟基自由基(・OH)的生成(方案 1a)。此外,所开发的基于皮克林乳液的光催化体系可在连续流模式下稳定运行,避免了分批反应中光催化剂的反复分离过程(方案 1b)。本文提出的利用皮克林乳液液滴界面调控活性氧物种生成浓度的策略,无需添加任何牺牲试剂,为碳氢键(C─H 键)的绿色氧化反应开辟了一条极具吸引力的新路径。
结果与讨论

图1.NC-3 光催化剂及皮克林乳液的表征结果。
基于皮克林乳液界面的光催化体系设计。
为构建基于皮克林乳液界面的光催化体系,需制备一种可紧密吸附在皮克林乳液液滴界面的两亲性光催化剂。选取溴氧化铋(BiOBr)作为光催化剂,因其铋 - 溴(Bi─Br)键键能较低,利于形成溴空位(VBr),从而促进分子氧的优先活化。采用厚度可控的二氧化硅(SiO₂)纳米片作为载体负载 BiOBr,所得负载型光催化剂自身可作为乳化剂,用于制备皮克林乳液。参照已报道的方法,通过延迟水解 - 缩合反应在 SiO₂纳米片的一侧修饰氨丙基,随后经辛基硅烷化处理,得到相对疏水的一侧。SiO₂纳米片未修饰的另一侧因存在大量羟基,具有相对亲水性。SiO₂纳米片上引入的氨丙基可与铋离子(Bi³⁺)发生配位作用,使溴氧化铋(BiOBr)仅负载于二氧化硅(SiO₂)纳米片的疏水侧。通过改变二氧化硅(SiO₂)前驱体的用量,制备出二氧化硅(SiO₂)纳米片厚度逐步递增的系列两亲性光催化剂,命名为 NC-n(n=1-4;n 代表纳米片厚度递增,对应厚度分别为 45、80、110 和 130 nm;厚度通过扫描电子显微镜(SEM),且溴氧化铋(BiOBr)均负载于二氧化硅(SiO₂)纳米片的疏水侧。
类似地,通过改变制备流程,可将氨丙基修饰在二氧化硅(SiO₂)纳米片的亲水侧或两侧。随后,采用上述相同的水热晶化法,制备出溴氧化铋(BiOBr)负载于 SiO₂纳米片亲水侧的 NC-3H 光催化剂(厚度 110 nm),以及 BiOBr 负载于 SiO₂纳米片两侧的 NC-3B 光催化剂(厚度 110 nm)。
以 NC-3 为代表性光催化剂进行表征分析。其透射电子显微镜(TEM)、高角环形暗场扫描透射电子显微镜(HAADF-STEM)及 X 射线能量色散谱(EDX)表征结果显示,不规则的溴氧化铋(BiOBr)薄片分布在二氧化硅(SiO₂)纳米片表面,横向尺寸为 2-4 微米(图 1a-g)。该观察结果与扫描电子显微镜(SEM)图像一致。由于 SiO₂纳米片一侧覆盖有 BiOBr 薄片,另一侧则相对光滑,结合胺基的配位作用可推断,BiOBr 仅负载于纳米片的疏水侧。电感耦合等离子体质谱(ICP-MS)测试结果表明,NC-3 光催化剂上 BiOBr 的负载量为 1.20 mmol/g。类似地,NC-3H 光催化剂中 BiOBr 分布于 SiO₂纳米片的亲水侧,而 NC-3B 光催化剂中 BiOBr 则分布于 SiO₂纳米片的两侧。此外,通过 X 射线衍射(XRD)、X 射线光电子能谱(XPS)、傅里叶变换红外光谱(FTIR)、紫外 - 可见漫反射光谱(UV-vis DRS)及莫特 - 肖特基(Mott-Schottky)分析对 NC-n(n=1-4)、NC-3H 及 NC-3B 光催化剂进行了全面表征。这些表征结果证实了各类光催化剂的成功制备。
如预期所示,所制备的两亲性光催化剂具有良好的皮克林乳液稳定能力(图 1h)。例如,以乙酸乙酯为油相,在 NC-3 光催化剂(相对于油相的质量分数为 7.7%)存在下,通过双相体系均化处理可制备出水包油(W/O)型皮克林乳液(平均液滴直径为 68 微米)。经水相和油相荧光染色验证,确认该乳液为 W/O 型皮克林乳液。此外,改变二氧化硅(SiO₂)纳米片的厚度或溴氧化铋(BiOBr)的负载位置,对皮克林乳液液滴尺寸无显著影响。将乳化后的双相体系经 24 小时光照后,皮克林乳液液滴的形貌仍保持完好。采用激光共聚焦扫描显微镜(CLSM)研究溴氧化铋(BiOBr)在皮克林乳液液滴界面的空间定位。二氧化硅(SiO₂)纳米片用异硫氰酸荧光素 I(FITC-I,发射绿色荧光)标记,而 BiOBr 自身在 405 nm 可见光激发下会产生蓝色荧光。对于 NC-3 光催化剂稳定的皮克林乳液体系,液滴界面周围出现了明显的荧光环(图 1i)。Z 堆叠三维 CLSM 图像显示,乳液液滴界面内侧(即水相近侧)仅出现绿色荧光,而 BiOBr 发射的蓝色荧光则位于皮克林乳液液滴界面外侧(即油相近侧)。这些结果表明,在水包油(W/O)型皮克林乳液体系中,BiOBr 可被定位在液滴界面外侧。当 SiO₂纳米片厚度从 45 nm 增加至 130 nm(对应 NC-1、NC-2、NC-4 光催化剂)时,蓝色荧光与绿色荧光内边缘的距离逐渐增大,意味着 BiOBr 与皮克林乳液液滴界面的距离越来越远(图 1j-l)。该观察结果证实,通过改变 SiO₂纳米片的厚度,可精准调控 BiOBr 在皮克林乳液液滴界面的空间位置。使用 NC-3H 光催化剂可实现 BiOBr 在皮克林乳液液滴界面内侧的定位,液滴界面内侧出现的蓝色荧光即为佐证。当 NC-3B 光催化剂用作乳化剂时,绿色和蓝色荧光峰几乎重叠,且液滴界面内侧和外侧均出现蓝色荧光,表明 BiOBr 分布在乳液液滴界面的两侧。此外,光催化反应进行 24 小时后,CLSM 图像显示 BiOBr 仍保持在皮克林乳液液滴界面的外侧、内侧或两侧,展现出良好的稳定性。
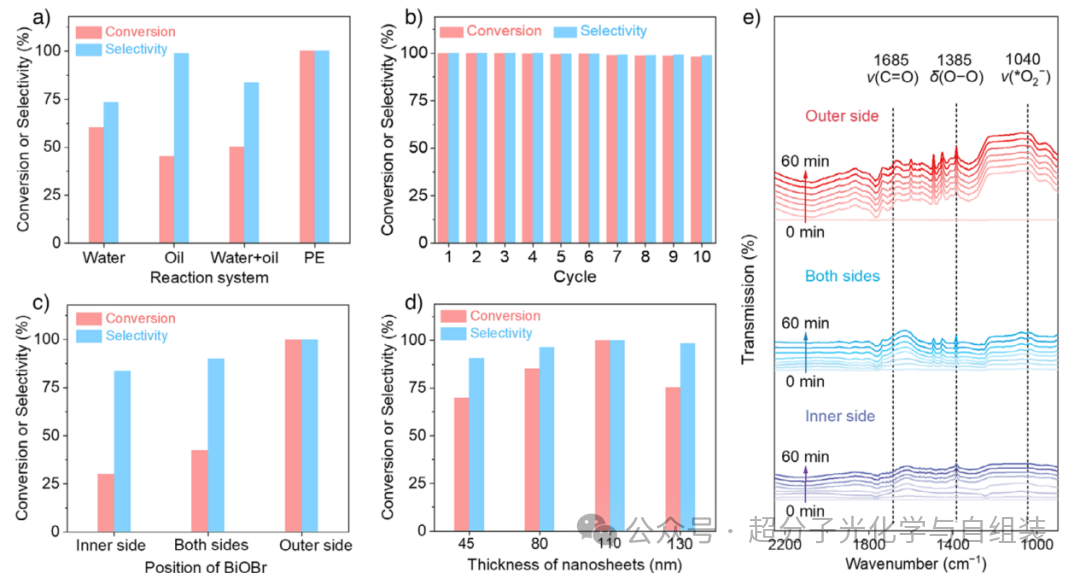
图2.皮克林乳液液滴界面及溴氧化铋(BiOBr)在界面的位置对乙苯光催化氧化反应的影响。。
皮克林乳液液滴界面的光催化选择性增强。
在对皮克林乳液液滴界面的光催化性能进行测试前,先分别研究了纯水相、纯油相及传统双相体系中的光催化活性。本研究筛选乙酸乙酯作为乙苯氧化反应的优选有机溶剂,并确定光源的最佳波长为 390 nm。光催化反应在经初步优化的反应条件下进行(LED 灯功率 20 W、光源波长 390 nm、反应温度 25 ℃)。如图 2a 所示,纯水相体系中乙苯的转化率达 60.2%,苯乙酮的选择性为 73.2%。在相同反应条件下,纯乙酸乙酯相体系中得到的乙苯转化率为 45.2%,而苯乙酮的选择性则大幅提升至 98.7%。将油相与水相混合后形成未乳化的传统双相体系,该体系中乙苯转化率为 50.2%、苯乙酮选择性为 83.6%,两项指标均介于纯水相与纯油相体系的测试结果之间。此外,二氧化硅(SiO₂)在纯水相、纯油相及传统双相体系中均未表现出任何光催化活性,而纯溴氧化铋(BiOBr)在这三种反应体系中展现的光催化活性与 NC-3 光催化剂相近。考虑到液 - 液界面面积的进一步增大或能凸显界面光催化的优势,上述结果促使作者开展基于皮克林乳液的界面光催化反应研究。不出所料,经 NC-3 光催化剂稳定的皮克林乳液体系实现了乙苯向苯乙酮的完全转化,乙苯转化率与苯乙酮选择性均高于 99.0%。这一结果表明,将光催化剂定位于皮克林乳液液滴界面,有利于提升乙苯光催化氧化反应的选择性。
分批反应结束后,吸附在液滴界面的 NC-3 光催化剂经高速离心实现回收。高速离心后对其进行洗涤、干燥处理。回收后的该光催化剂仍可乳化双相体系,形成皮克林乳液。将该回收的 NC-3 光催化剂继续用于后续九次催化反应,乙苯转化率仍>98.0%,苯乙酮选择性仍>99.0%(图 2b)。此外,经十次循环反应后,由 NC-3 光催化剂稳定的皮克林乳液液滴的激光共聚焦扫描显微镜图像显示,溴氧化铋仍保持在液滴界面外侧的空间定位。多种表征手段(X 射线衍射、X 射线光电子能谱、扫描电子显微镜、透射电子显微镜)证实,NC-3 光催化剂在光照条件下具备良好的稳定性。皮克林乳液体系中的 NC-3 光催化剂表观量子产率为 2.2%,远高于水相反应体系中经晶格工程改性的溴氧化铋光催化剂 —— 即便在叔丁醇存在的条件下,该改性光催化剂的表观量子产率仅为 1.55%(对应乙苯转化率 95.0%、苯乙酮选择性 98.0%)。与叔丁基过氧化氢介导的氧化体系相比,本研究构建的基于皮克林乳液的光催化体系展现出更优异的反应活性与选择性;且因体系中无氧化剂衍生的其他副产物生成,还能简化产物的分离与纯化流程。此外,苯乙酮的这一超高选择性优于已报道的光催化或热催化体系相关结果。
溴氧化铋的负载位置对反应选择性的影响。
为进一步探究界面效应,作者通过精准调控溴氧化铋(BiOBr)在皮克林乳液液滴界面的位置,开展了多组光催化反应测试。首先,在相同反应条件下,对 BiOBr 分别位于皮克林乳液液滴界面外侧、内侧及两侧的三种体系进行了光催化反应性能评价。结果表明,BiOBr 位于皮克林乳液液滴界面外侧的体系中,乙苯转化率与苯乙酮选择性均高于 99.0%,显著优于 BiOBr 位于界面内侧(转化率 30.0%、选择性 83.6%)和界面两侧(转化率 42.4%、选择性 90.1%)的体系(图 2c)。本研究采用原位漫反射红外傅里叶变换(DRIFT)光谱对光催化反应过程进行了实时监测(图 2e)。在 60 分钟的原位漫反射红外傅里叶变换光谱连续扫描测试中,这三种反应体系均在 1683 cm⁻¹ 处出现特征吸收峰,该峰归属于苯乙酮分子中羰基(C═O)的伸缩振动。值得注意的是,在整个光催化反应过程中,溴氧化铋(BiOBr)位于乳液液滴界面外侧的反应体系中,该羰基特征峰的强度为三种体系中最强。这一实验结果证实,将 BiOBr 定位于皮克林乳液液滴界面外侧,有利于实现乙苯的光催化选择性氧化。
此外,由于二氧化硅(SiO₂)纳米片厚度越大,溴氧化铋(BiOBr)与油水界面的距离就越远,通过调控 SiO₂纳米片的厚度,精准调节了 BiOBr 在乳液液滴界面外侧的具体位置,并对其光催化活性进行了测试。如图 2d 所示,当 SiO₂纳米片厚度为 45 nm 时,乙苯转化率为 69.7%,苯乙酮选择性为 90.5%。有趣的是,当 SiO₂纳米片厚度逐步增加至 110 nm 时,乙苯几乎完全被氧化为苯乙酮,苯乙酮选择性高于 99.0%。但当 SiO₂纳米片厚度进一步增至 130 nm 时,乙苯的转化率和苯乙酮的选择性均开始下降,分别降至 75.2% 和 98.4%。上述结果清晰表明,溴氧化铋(BiOBr)在皮克林乳液液滴界面外侧的空间定位,对光催化反应的结果具有显著影响。
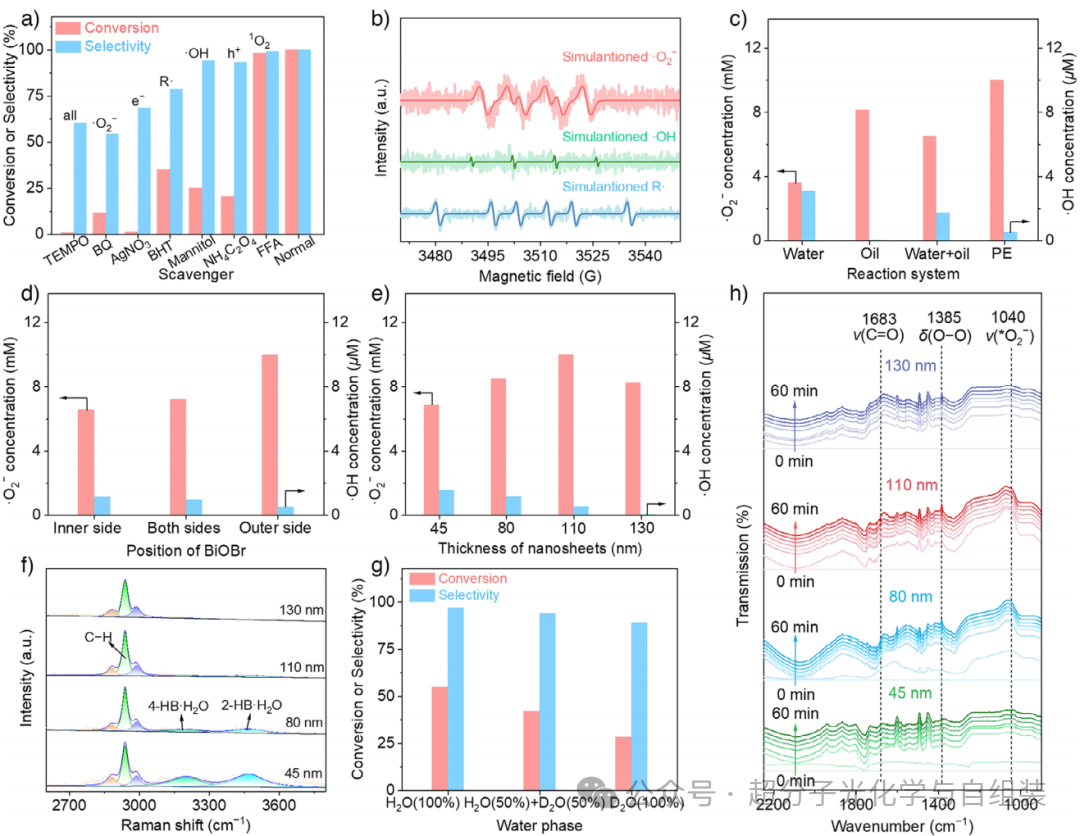
图3.皮克林乳液液滴界面生成的超氧阴离子自由基(・O₂⁻)和羟基自由基(・OH)浓度与乙苯光催化氧化反应选择性的关系。
选择性提升的原因探析。
为探究不同类型活性氧物种在乙苯光催化氧化反应中的作用,开展了一系列自由基捕获实验(图 3a)。加入 2,2,6,6 - 四甲基哌啶氮氧自由基(TEMPO)作为猝灭剂后,反应体系的催化效果发生了相应变化。催化活性几乎完全丧失。停止光照后,光催化活性急剧下降,但反应并未完全终止,这是由于反应体系中残留的活性氧物种仍能继续推动氧化反应进行;恢复光照后,反应则恢复至原有水平。上述结果证实,该氧化反应为自由基引发型反应。加入 1,4 - 苯醌(BQ)作为超氧阴离子自由基(・O₂⁻)捕获剂后,实验观察到乙苯转化率从 99.0% 以上骤降至 11.7%,苯乙酮选择性也从 99.0% 以上降至 54.6%,这表明・O₂⁻是实现乙苯氧化生成苯乙酮的关键活性氧物种。当硝酸银(AgNO₃)对光生电子(e⁻)进行捕获时,反应几乎完全停止,这表明超氧阴离子自由基(・O₂⁻)来源于分子氧被光生电子还原的过程。采用 2,6 - 二叔丁基 - 4 - 甲基苯酚(BHT)猝灭碳中心自由基(R・)后,乙苯转化率出现显著下降,证实碳中心自由基是影响乙苯转化的重要活性物种。加入甘露醇或草酸铵(NH₄C₂O₄)时,乙苯转化率虽大幅降低,但苯乙酮的选择性仍保持在较高水平,这意味着光生空穴(h⁺)氧化水分子生成的羟基自由基(・OH)会促进碳中心自由基的生成。使用糠醇(FFA)捕获单线态氧(¹O₂)后,体系的光催化活性几乎未发生变化,由此排除了单线态氧介导该氧化反应的可能性。在该光催化体系中通入氮气营造无氧氛围后,乙苯转化率从 55.0% 降至 8.9%。随后,分别采用 ¹⁸O₂和 H₂¹⁸O 开展氧 - 18 同位素标记实验,探究反应过程中氧原子的迁移路径。实验结果表明,源于分子氧的超氧阴离子自由基是该光催化氧化反应的关键氧源 —— 在 ¹⁸O₂参与的反应体系中,质谱检测发现氧 - 18 被引入至苯乙酮分子中。本研究采用电子顺磁共振(EPR)光谱对光催化过程中生成的活性物种进行检测,成功捕捉到对应超氧阴离子自由基、羟基自由基和碳中心自由基的特征信号(图 3b),证实了这三种活性物种在光催化反应过程中均存在。
分别以氯化硝基四氮唑蓝和香豆素为探针,对光催化过程中超氧阴离子自由基(・O₂⁻)和羟基自由基(・OH)的浓度进行了定量检测。如图 3c 所示,纯水相体系中检测到的・O₂⁻和・OH 浓度分别为 3.6 mM 和 3.1 μM;纯油相体系中则仅生成・O₂⁻,且其浓度高达 8.1 mM。与水相比,有机溶剂对分子氧具有更好的溶解性,有利于・O₂⁻的生成,但因体系中缺乏水,无法发生氧化反应生成・OH。相较于纯水相体系,传统双相体系中的・O₂⁻浓度提升至 6.5 mM,而・OH 浓度则降至 1.7 μM,这表明液 - 液双相界面会对・OH 的生成产生影响。随后,进一步探究了皮克林乳液体系中・O₂⁻和・OH 的生成浓度:当 BiOBr 定位于皮克林乳液液滴界面内侧时,体系中・O₂⁻和・OH 的浓度分别为 6.6 mM 和 1.1 μM(图 3d);与之相反,当 BiOBr 分布在乳液液滴界面两侧时,体系中・O₂⁻浓度升至 7.2 mM,・OH 浓度则降至 1.0 μM。值得注意的是,当 BiOBr 定位于乳液液滴界面外侧时,皮克林乳液体系中的・O₂⁻浓度大幅提升至 10.0 mM,该数值甚至高于纯油相体系中的・O₂⁻浓度,而体系中・OH 的浓度则低至 0.5 μM。此外还进一步研究了二氧化硅(SiO₂)纳米片的厚度对体系中・O₂⁻和・OH 生成浓度的影响。当二氧化硅(SiO₂)纳米片的厚度从 45 nm 逐步增加至 110 nm 时,光催化体系中的超氧阴离子自由基(・O₂⁻)浓度由 6.9 mM 升高至 10.0 mM(图 3e);而当纳米片厚度进一步增加至 130 nm 时,反应体系中的・O₂⁻浓度则开始下降,降至 8.2 mM。值得关注的是,随着 SiO₂纳米片厚度的增加,羟基自由基(・OH)的浓度从 1.5 μM 逐步降低至 0.5 μM,当 SiO₂纳米片的厚度达到 130 nm 时,光催化体系中几乎检测不到・OH 的存在。上述结果表明,将溴氧化铋(BiOBr)精准定位于皮克林乳液液滴界面外侧,不仅有利于・O₂⁻的生成,还能抑制・OH 的产生。
为阐明 BiOBr 处于不同位置时体系中・O₂⁻和・OH 浓度发生变化的原因,结合界面水与・OH 的生成密切相关这一特性,将激光光斑移至皮克林乳液液滴边缘,采用原位拉曼光谱对界面处的水分子信息进行了捕获分析。如图 3f 所示,位于 3200 cm⁻¹ 和 3470 cm⁻¹ 处的特征峰分别归属于高配位氢键水(4-HB・H₂O,体相水)和低配位氢键水(2-HB・H₂O,界面水)。当纳米片厚度为 45 nm 的 NC-1 光催化剂组装于乳液液滴界面时,在 BiOBr 所处的乳液液滴界面外侧,主要存在 4-HB・H₂O 和 2-HB・H₂O 两种形态的水。随着纳米片厚度的增加,定位于乳液液滴界面外侧的 BiOBr 与水相间的距离逐渐增大,4-HB・H₂O 和 2-HB・H₂O 对应的特征峰强度均呈下降趋势;当纳米片厚度增至 110 nm 时,体系中仅能检测到 2-HB・H₂O 的特征峰;而当纳米片厚度进一步增至 130 nm 时,水分子的特征信号几乎完全无法检测。这表明,随着 SiO₂纳米片厚度的增加,乳液液滴界面处的水密度逐渐降低,氢键作用也随之变弱。此外,还通过原位漫反射红外傅里叶变换光谱,利用 O─O 键在 1385 cm⁻¹ 处的转动振动特征峰,对 NC-n(n=1-4)系列光催化剂表面的分子氧吸附行为进行了测定(图 3h)。当纳米片厚度从 45 nm 增加至 110 nm(对应 NC-1、NC-2、NC-3 光催化剂)时,O─O 键的特征峰强度逐渐增强,这主要是因为 BiOBr 逐渐向油相区域延伸,而油相中富氧的亲脂性溶剂化环境有利于分子氧的化学吸附,进而促进生成更高浓度的・O₂⁻(吸附态超氧物种O₂⁻在 1040 cm⁻¹ 处有特征信号)。当纳米片厚度增至 130 nm(对应 NC-4 光催化剂)时,体系中O₂⁻的特征峰强度出现下降,这是因为 BiOBr 周边缺乏水分子来消耗生成的光生空穴 h⁺,最终导致了电子 - 空穴对的复合。
综合上述各项实验结果,可以推测:皮克林乳液液滴界面外侧的氢键作用逐渐减弱,这一情况不利于羟基自由基(・OH)的生成。为验证该假说,设计对照实验,在水相中加入重水(D₂O),以增强 NC-3 光催化剂稳定的乳液液滴界面处的氢键作用。随着水相中重水占比从 0 提升至 100%,氢键作用逐渐增强,乙苯的转化率(从 55.0% 降至 28.3%)和苯乙酮的选择性(从 97.0% 降至 89.1%)均随之下降(图 3g)。此外,体系中超氧阴离子自由基(・O₂⁻)的浓度也从 10.0 mM 降至 6.7 mM。这是因为液滴界面处氢键作用的增强会降低分子氧的传质系数,进而削弱分子氧的化学吸附作用,导致・O₂⁻生成浓度降低,这一结论也得到了原位漫反射红外傅里叶变换光谱的验证。有趣的是,加入重水后,体系中・OH 的浓度从 0.5 μM 升至 0.9 μM。这一结果明确证实,液滴界面处氢键强度的变化会影响・OH 的生成。在 NC-1 光催化剂稳定的光催化乳液体系中,作者也观察到了类似现象,说明通过调控液滴界面处的氢键作用来调节活性氧物种的生成,是一种具有普适性的方法。上述实验结果为以下结论提供了有力证据:乳液液滴界面外侧独特的弱氢键环境,既有利于・O₂⁻的生成,又能抑制・OH 的产生,从而实现乙苯向苯乙酮的高效选择性转化。
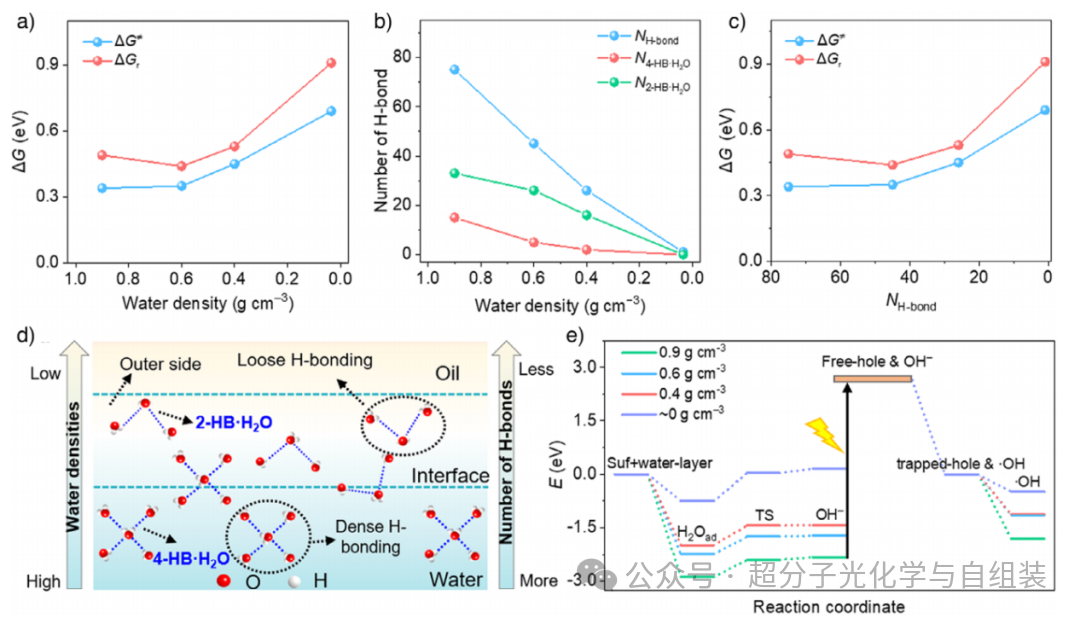
图4.密度泛函理论(DFT)计算结果。
作者采用密度泛函理论(DFT)和从头算分子动力学(AIMD)模拟,探究了皮克林乳液液滴界面在不同水密度条件下羟基自由基(・OH)的生成规律。模拟结果表明,水分子的解离过程涉及单个或多个水分子的参与。在水密度足够高时,解离能垒(ΔG≠)的变化幅度极小(水密度为 0.9 g・cm⁻³ 时解离能垒为 0.34 eV,0.6 g・cm⁻³ 时为 0.35 eV);而当水密度从 0.6 g・cm⁻³ 降至接近 0 g・cm⁻³ 时,解离能垒则呈逐步升高趋势,从 0.35 eV 升至 0.69 eV(图 4a)。在高水密度条件下(0.9 和 0.6 g・cm⁻³),反应自由能(ΔGr)的数值相近,分别为 0.49 eV 和 0.44 eV;当水密度降至接近 0 g・cm⁻³ 时,反应自由能则逐步升高至 0.91 eV。此外,还探究了氢键数量与水密度之间的关联,发现当水密度逐步降至接近 0 g・cm⁻³ 时,氢键总数(NH-bond)、高配位氢键水的氢键数(N4-HB-H₂O)以及低配位氢键水的氢键数(N2-HB-H₂O)均呈下降趋势(图 4b)。这三个参数均与解离能垒(ΔG≠)和反应自由能(ΔGr)呈正相关关系(图 4c)。羟基自由基(・OH)的生成过程分为两个步骤:(1)水分子解离生成氢氧根离子(OH⁻);(2)氢氧根离子与表面空穴相互作用,生成羟基自由基(・OH)。由此可以推断,由低水密度引发的弱氢键作用,会通过提高解离能垒(ΔG≠)和反应自由能(ΔGr),抑制水分子解离生成氢氧根离子(OH⁻),进而抑制羟基自由基(・OH)的生成。当水密度从 0.9 g・cm⁻³ 降至接近 0 g・cm⁻³ 时(图 4d),水层与 H₂O/BiOBr 之间的相互作用能(Eint)从 - 2.88 eV 降至 - 0.75 eV,水层与・OH/BiOBr 之间的相互作用能(Eint)也从 - 1.81 eV 降至 - 0.49 eV(图 4e)。这表明较高密度的水能够稳定整个体系,从而稳定生成的羟基自由基(・OH)。上述理论计算结果与实验测得的活性氧物种浓度变化规律一致。综上,(1)皮克林乳液液滴界面外侧低水密度导致的弱氢键作用,会提高水分子的解离能垒,不利于羟基自由基(・OH)的生成;(2)皮克林乳液界面可营造富氧的亲脂性溶剂化环境,促进分子氧的化学吸附,加速超氧阴离子自由基(・O₂⁻)的生成。
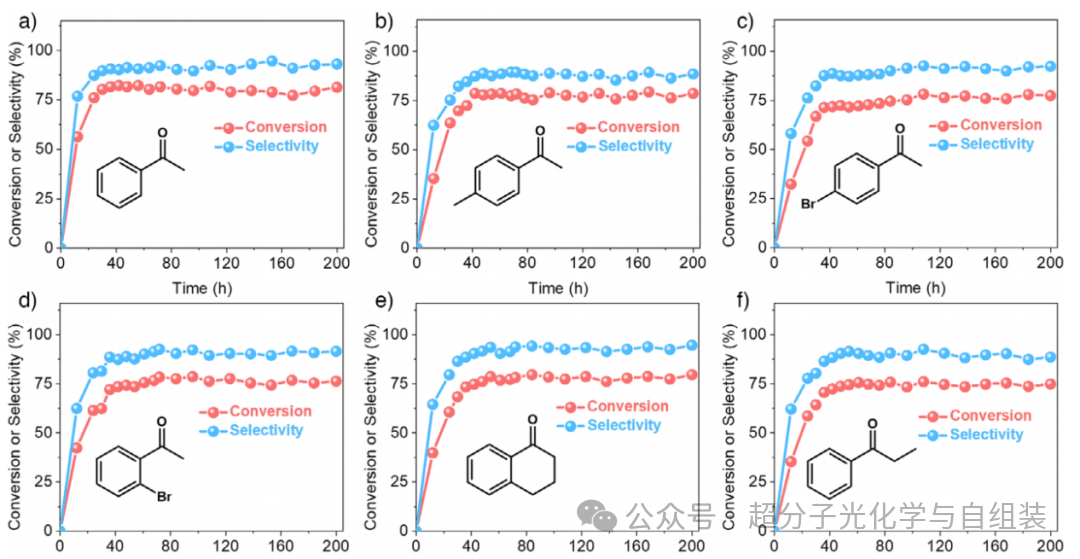
图5.乙苯及其他芳香族 C—H 键的连续流光催化氧化反应结果。
尽管皮克林乳液液滴界面外侧的光催化氧化活性得到了提升,但分批式光催化反应仍存在两个局限性:(1)每批反应结束后需破乳才能分离光催化剂;(2)釜式反应器中光的穿透深度有限,难以实现光催化过程的放大。本课题组提出的基于皮克林乳液的连续流反应体系,有望解决这些问题。采用与分批反应相同的方法制备水包油(W/O)型皮克林乳液,将其注入柱式反应器中。反应器底部装有微米级孔径的过滤器(孔径小于乳液液滴尺寸),可使乳液液滴在反应器内堆积且不渗漏。氧气从反应器顶部通入,圆柱形 LED 光源从四周对柱式反应器进行照射。
如图 5a 所示,连续流反应 48 h 后,乙苯转化率约为 81.6%,苯乙酮选择性约为 92.3%。在 200 h 的光催化氧化过程中,这两个数值均保持良好,仅出现微小波动。长时间运行后,由于层状光催化剂在乳液液滴界面的强吸附作用以及表面润湿性变化较小,皮克林乳液的液滴尺寸未发生显著增大。此外,还探究了该乳液液滴界面光催化氧化体系的底物适用范围。含给电子基团的芳基烷烃(如对甲基乙苯)表现出优异的转化率(约 77.9%)和对相应酮类产物的选择性(约 88.9%)(图 5b)。以对溴乙苯为底物时,连续流反应 48 h 后获得了中等转化率(约 72.4%)和选择性(约 87.6%)(图 5c),这归因于对位的吸电子基团。当以邻溴乙苯为底物时,光催化反应的转化率和选择性分别提升至 74.2% 和 88.9%(图 5d)。此外,该光催化剂对四氢萘和正丙苯的选择性氧化也表现出良好的催化性能(图 5e~f)。连续流反应的转化率和选择性略低于分批反应,这可能是由于位于柱式反应器中部的乳液液滴无法充分接收光照所致。未来可通过减小柱径、延长柱长来克服这一限制。总之,本研究首次实现了基于皮克林乳液界面的连续流光催化合成。
文章总结
本研究成功开发了一种利用皮克林乳液液滴界面增强 C─H 键光催化选择性氧化的策略。其核心在于将溴氧化铋(BiOBr)光催化剂精准定位于水包油(W/O)型皮克林乳液液滴的外侧界面,该位置有利于目标活性氧物种的生成。因此,在室温、可见光照射下,乙苯可高选择性地转化为苯乙酮。其转化率和选择性显著高于乳液液滴界面内侧、两侧、传统双相体系、纯水相及纯油相中的反应结果。乙苯光催化氧化选择性的显著提升,归因于界面处的弱氢键作用 —— 该作用提高了水分子的解离能垒和反应自由能,从而抑制羟基自由基(・OH)的生成;同时,界面体系营造的富氧亲脂性溶剂化环境,促进了分子氧的化学吸附,加速超氧阴离子自由基(・O₂⁻)的生成。此外,该界面光催化体系可实现连续流反应,具有高活性和长期运行稳定性(200 h)。本研究提出的 “利用皮克林乳液液滴界面调控活性氧物种生成(无需牺牲剂)并构建连续流光催化体系” 的策略,为提高光催化选择性乃至光催化技术的实际应用开辟了新路径。
一 审:张雅蓉
二 审:马俊红
三 审:郭 炜









